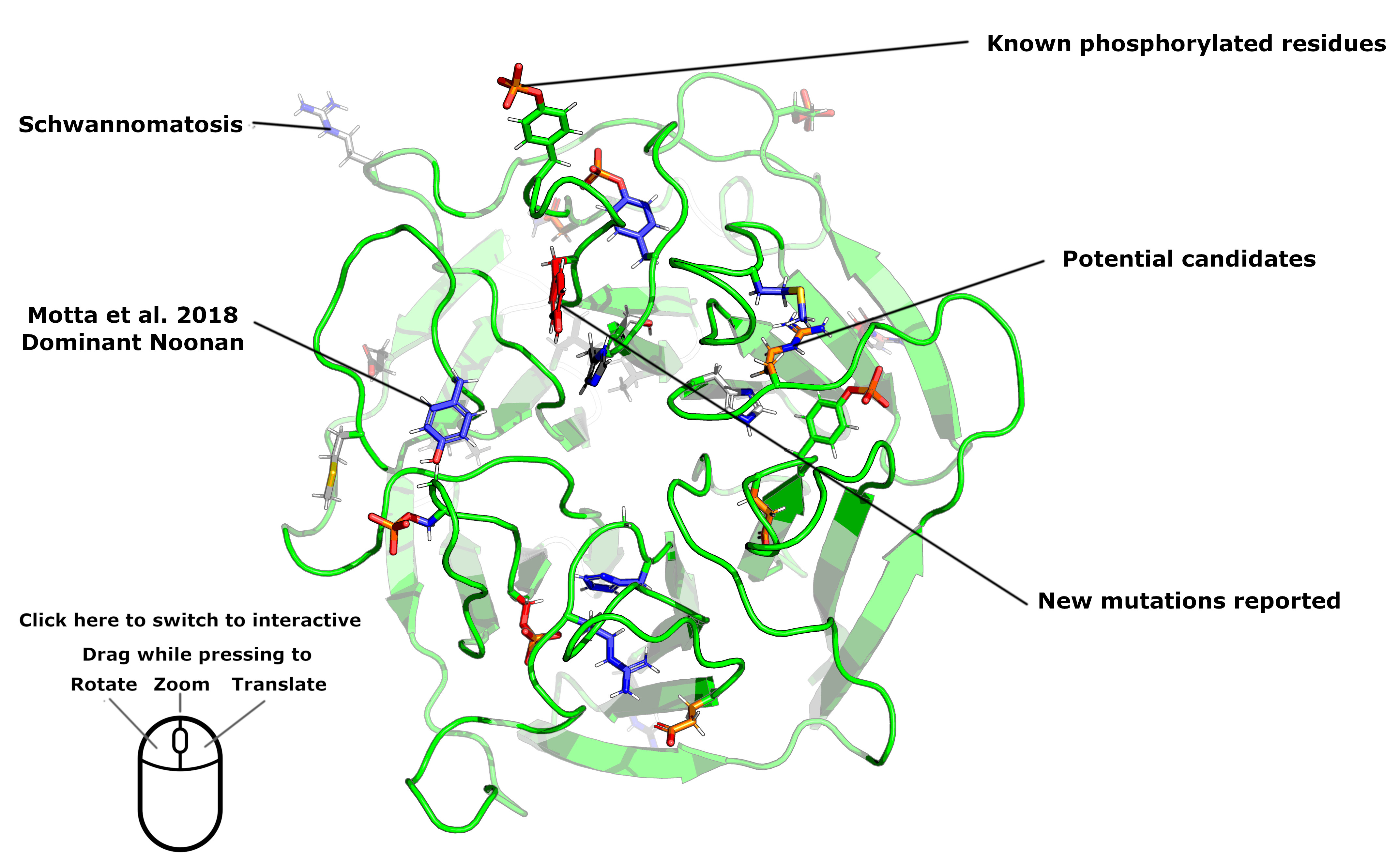
A recent study by Motta et al performed homology modelling on the LZTR1 protein and showed that the kelch (KT) domains resemble a six-bladed propeller like structure. It was further shown that autosomal dominant acting mutations typically lie in the upper surface of this model. In order to show where the autosomal dominant acting mutations identified in the present study lay, two structural homology models were made. The first was with I-TASSER using threading and ab initio construction with evolutionary constraints. To more closely replicate the work of Motta et al, a second model was made with Phyre2
in one-to-one threading mode. In this second model, a 1.4Å X-ray diffraction structure of Ta-TFP
(a thiocyanate-forming protein) from Thlaspi arvense was used as a template (PDB 5A10). PhosphoSite Plus
was queried for known phosphorylation sites and these residues were modified in the models by changing the relevant amino acid codes (SER>SEP, THR>TPO, TYR>PTR) and correcting/energy minimising the model with Rosetta Relax.
The structures were examined with PyMol 2.2 (Schrödinger Inc.).
As the second model was better structured, we present results from that model (see image below) and show the position of the 5 de novo and one inherited AD-acting mutations. As shown, 4 of the 6 variants identified lie on the upper side of this propeller. The exceptions are p.R97L and p.N145I which both appear to be buried toward the side of the propeller. We note that many of the AD-associated variants lie in close proximity to, or are themselves, known phosphorylated residues. In particular, p.S244C disrupts a known phosphorylation site and is unlikely to disrupt hRAS binding due to the amino-acid change alone.
The hypothesis that disruption of phosphorylation may affect protein interaction may be supported by a comparison to KLHL3, a gene that encodes a homologous kelch-domain protein. Mutations in this gene have been linked to hypertension and electrolyte abnormalities. Although families with both dominant and recessive modes of inheritance were described, it was noted that dominant-acting mutations typically cluster in short segments within the propeller-like structure.
A more detailed structural analysis went on show that phosphorylation of serine 433 impeded substrate-binding at the interface, whereas a protein harbouring the dominant, disease-associated p.S433N could not be repressed by this mechanism.
The relative positions of candidate LZTR1 heterozygous variants (including p.N145I) identified in singleton patients from the DDD study are documented in Table S3 and shown in the figure below (orange). Green denotes wild-type residues. Blue side chains show the known dominant-acting variants described in Motta et al.4 Red side chains highlight residues disrupted by de novo mutations identified as part of the current study.